| Issue |
Vis Cancer Med
Volume 6, 2025
|
|
|---|---|---|
| Article Number | 9 | |
| Number of page(s) | 11 | |
| DOI | https://doi.org/10.1051/vcm/2025010 | |
| Published online | 24 July 2025 | |
Perspective Article
Tumor-infiltrating lymphocyte therapy: therapeutic advances and prospects
1
Department of Chemistry and Biochemistry, Florida State University, Tallahassee, FL 32306-4390, USA
2
Institute of Molecular Biophysics, Florida State University, Tallahassee, FL 32306-4390, USA
3
Institute for Pediatric Rare Diseases, Florida State University, Tallahassee, FL 32306-4390, USA
* Corresponding authors: This email address is being protected from spambots. You need JavaScript enabled to view it.
(D.B. Sarker); This email address is being protected from spambots. You need JavaScript enabled to view it.
(Q.-X.A. Sang)
Received:
13
May
2025
Accepted:
30
June
2025
Abstract
Tumor-infiltrating lymphocyte (TIL) therapy has evolved from a pioneering experimental approach to a clinically validated treatment strategy, underscored by the recent approval of lifileucel (Amtagvi) by the Food and Drug Administration (FDA) for advanced melanoma refractory to existing therapies. Initially successful in melanoma due to its high tumor mutational burden (TMB) and immune-reactivity, contemporary efforts extend TIL applications to other solid tumors, including lung, cervical, and colorectal cancers. However, these lower-TMB malignancies typically require the selective enrichment of tumor-specific T cells to achieve significant clinical efficacy. The therapeutic potential of TILs is influenced by critical factors, including cell dose, T-cell phenotype and differentiation state, tumor-specific reactivity, and the ability to persist and expand within patients post-infusion. Emerging techniques, including single-cell transcriptomics and biomarker-guided TIL selection (e.g., CD137, CD103 markers), have provided deeper insights into the characteristics correlating with successful outcomes. Ongoing clinical trials highlight future directions, including genetically engineered TILs with chimeric antigen receptor (CAR) or immune checkpoint knockout, improved cytokine support strategies to enhance T-cell expansion and reduce toxicity, and optimized lymphodepletion regimens. Establishing clear quality attributes for TIL manufacturing will be essential for consistent clinical success, paving the way toward personalized and robust immunotherapeutic approaches across diverse cancer types.
Key words: Tumor-infiltrating lymphocytes / TIL therapy / Immunotherapy / Adoptive cell therapy
© The Authors, published by EDP Sciences, 2025
 This is an Open Access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Introduction
Tumor‐infiltrating lymphocyte (TIL) therapy is a personalized immunotherapy modality that harnesses a patient’s own tumor‐resident T cells, which are isolated from resected tumor specimens, expanded ex vivo under precise culture conditions, and subsequently reinfused to mount a targeted antitumor response (Figure 1) [1–4]. Initially developed in the 1980s through pioneering work by Rosenberg and colleagues [5], early clinical trials in metastatic melanoma demonstrated that IL-2-expanded TILs could induce durable tumor regressions in patients with high mutational burdens and abundant neoantigens, providing early proof‐of‐concept for this adoptive cell transfer approach [2, 6, 7]. Recent clinical advances have extended the application of TIL therapy beyond melanoma to other solid tumors, including non-small cell lung cancer (NSCLC), head and neck squamous cell carcinoma (HNSCC), cervical carcinoma, breast cancer, colorectal cancer, and osteosarcoma [8]. A key milestone in this evolution was achieved in February 2024 when the US Food and Drug Administration (FDA) granted accelerated approval to lifileucel (Amtagvi), the first autologous TIL product and the first cellular therapy approved for a solid tumor indication, thereby validating TIL therapy as a clinically viable treatment option [9, 10]. Lifileucel is approved for adults with advanced melanoma refractory to standard immunotherapies, based on a pivotal clinical trial in which 31.4% of patients (48 out of 153 across two cohorts) achieved an objective response, including 8 complete responses and 40 partial responses. [11]. This success has sparked intensified research into TIL therapy for other malignancies, with ongoing trials investigating its use in cancers with traditionally low immunogenicity as well as in those with established viral etiologies [12].
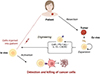 |
Figure 1 Overview of tumor-infiltrating lymphocyte (TIL) therapy. Tumors are surgically resected from patients, and tumor-infiltrating lymphocytes (TILs) are isolated and expanded ex vivo. These cells may either be infused without modification or genetically engineered. Engineered TILs include CAR (chimeric antigen receptor) TILs, checkpoint protein knockout (KO) TILs (e.g., programmed cell death protein 1 (PD-1) KO), and TILs expressing cytokine (e.g., interleukin-15 or IL-15) or chemokine receptor (e.g., CXC chemokine receptor 2 or CXCR2) to enhance persistence, function, or trafficking. Upon reinfusion into the patient, activated TILs specifically home to and target cancer cells, mediating effective tumor cell killing and potentially leading to durable cancer regression. |
Mechanistically, TILs function by their diverse T cell receptor (TCR) repertoire, enabling simultaneous recognition of multiple tumor-associated antigens and driver neoantigens, thereby overcoming the limitations of monovalent approaches like chimeric antigen receptor (CAR)-T therapies in heterogeneous solid tumors [2, 6]. Their antitumor efficacy is driven by direct cytolytic activity mediated via perforin and granzyme secretion, as well as by the production of cytokines such as interferon-gamma (IFN-γ) that help reshape the immunosuppressive tumor microenvironment [13, 14]. However, the manufacturing process for TILs remains laborious and time-consuming, often requiring a 3- to 6-week period for ex vivo expansion and selection [15, 16], which may delay treatment for patients with rapidly progressing disease. Moreover, the immunosuppressive tumor microenvironment – including the presence of regulatory T cells, myeloid-derived suppressor cells, and high levels of inhibitory cytokines – can attenuate the efficacy of reinfused TILs [6, 17]. To overcome these obstacles, current research is combining TIL therapy with checkpoint inhibitors and immunomodulators [18, 19], while also using genetic engineering, such as knockout of programmed cell death protein 1 (PD-1) [20] or transduction of interleukin-15 (IL-15) [21] or CXC motif chemokine receptor 2 (CXCR2) [22] genes – to enhance T cell persistence and antitumor activity.
In this perspective article, we discuss the therapeutic successes of TIL therapy and outline the current framework and product attributes underpinning these outcomes. We further highlight insights gained from clinical trials that may help refine existing approaches and explore promising next-generation TIL therapies poised to further enhance clinical outcomes.
Evolution of TIL therapy across tumor types
TIL therapy’s evolution reflects a contrast between its promise and the challenges of different tumor environments. After the initial proof-of-concept in melanoma, efforts to implement TIL therapy to other malignancies progressed slowly. Melanoma’s high tumor mutational burden (TMB) and abundant T-cell infiltrates meant that even “bulk” TIL cultures (expanded en masse without specific selection) could contain tumor-reactive lymphocytes capable of mediating remission. In contrast, many epithelial cancers with lower TMB appeared “colder” – fewer neoantigens and often a more immunosuppressive microenvironment – yielding lower response rates to unselected TIL therapy. Consequently, for many years, meaningful clinical successes were largely confined to melanoma.
However, the past decade has seen renewed progress in extending TIL therapy beyond melanoma. Studies have demonstrated that even lower-TMB solid tumors can respond to TIL therapy if the TILs are enriched for the right specificities. For example, in non-small cell lung cancer (NSCLC), a recent trial successfully expanded tumor-reactive TIL cultures from 18 of 20 patients, treating 16 of them and achieving durable complete remissions in 2 patients [23]. Another striking proof-of-concept came from a cervical carcinoma study where HPV E6- and EC7-reactive TIL infusion yielded complete responses in 2 of the 9 treated patients [24]. However, detailed analysis revealed that T cells driving long-term benefit were not specific for the expected HPV viral oncoprotein; instead, they targeted unique neoepitopes or a cancer germline antigen [25]. Still, having HPV-reactive T cells in the final product was associated with clinical benefit, and the protocol was refined to require a minimum number of HPV-specific TIL cultures for treatment [26]. These findings illustrate that even in tumors with an obvious shared antigen (e.g., viral proteins), the key is to identify truly effective clones that may target patient-specific neoantigens. Durable complete responses often lasting >4 years have been achieved in most trials once the correct TIL specificities were isolated. In a pilot study involving 18 patients with gastrointestinal cancers, treatment with unselected TILs failed to induce any objective clinical responses [27]. However, retreatment of one patient using a TIL product containing approximately 95% neoantigen reactive cells resulted in a partial response in liver and lung metastases that was sustained for 34 months [28]. Together, these studies underscore a critical insight: melanoma and other high-TMB cancers may be treated with bulk TILs, whereas low-TMB tumors usually demand a more tailored approach, enriching for tumor-specific lymphocytes to attain meaningful responses. This evolution from bulk to selected TIL products defines the current landscape as TIL therapy spreads to diverse cancers.
Factors determining TIL product quality and clinical efficacy
The clinical success of TIL therapy hinges on multiple biological and manufacturing factors that affect the potency of the infused lymphocytes. Key determinants include their phenotypic and functional qualities, their specificity for tumor antigens, the number of cells infused, and the supportive therapies given alongside the TILs. Recent research and clinical experience have shed light on the attributes of a TIL product and the manufacturing process that are most critical for driving tumor regression.
TIL phenotype and differentiation state
TIL products primarily contain T-cells characterized by the expression of CD45 and CD3 markers [29]. Among these T-cells, clinical efficacy has been strongly associated with elevated proportions of cytotoxic CD8+ T-cells [30, 31]. Nonetheless, helper CD4+ T-cells, which also exhibit tumor-specific reactivity in vitro, likely play an important supportive role [32]. The differentiation and exhaustion status of TILs profoundly influences their therapeutic efficacy. Effector CD8+ T-cells within the tumor frequently display features of exhaustion, marked by elevated expression of inhibitory receptors (PD-1, TIM-3, LAG-3), along with surface markers like CD39 phenotype [33, 34]. These chronically stimulated cells can have poor proliferative capacity and functionality. In contrast, within the tumor and lymph nodes, there exists a minority of “stem-like” effector and memory CD8+ T cells that retain robust proliferative potential and multipotency (longer telomeres, often marked by TCF-1 expression, co-expression of costimulatory receptors like CD28, and absence of terminal exhaustion markers like TIM-3 and CD39) [35, 36]. Data from TIL therapy trials indicate that products containing a higher proportion of such stem-like or early memory T cells are associated with better outcomes [37–39]. One study showed these stem-like TILs were 2.5-fold more abundant in patients with complete responses compared to non-responders, and patients with a higher frequency of CD39−CD69− T cells in the infusion had significantly improved overall survival [9]. Conversely, overly differentiated effector TILs, while highly cytotoxic initially, may lack the persistence to be tumor-reactive when infused back to the patient [40]. Interestingly, there is some nuance – one group observed that an infusion product enriched in robustly cytotoxic effector CD8+ T cells with strong tissue-homing markers correlated with better outcomes [30]. This suggests that both qualities – sufficient effector function and long-term persistence – are desirable. Achieving the right balance in TIL phenotype is an active area of optimization. In practice, shorter ex vivo culture times (the “Young TIL” approach) tend to yield a less-differentiated product with improved persistence after infusion [39, 41, 42].
Tumor specificity and reactivity
A critical factor is what fraction of the infused TILs are actually tumor-reactive (i.e., recognize tumor antigens) as opposed to being “bystanders.” Bulk TIL cultures from a tumor naturally contain a mix of T cells, not all of which are specific for the cancer – some may be specific for viruses or other antigens unrelated to the tumor’s malignant cells. The clinical potency of a TIL product stems from the subset that can recognize and kill tumor cells. Thus, identifying and expanding these tumor-reactive lymphocytes is pivotal. In manufacturing, this is often assessed by assays such as IFN-γ release when TILs are co-cultured with the patient’s tumor. For instance, one trial defined a “successful” TIL product by a minimum IFN-γ secretion threshold (e.g., ≥400 pg/mL in at least one of the culture wells) and found that using this criterion, tumor-reactive TILs could be grown from 67% of patient tumors [9]. Tumor samples vary – those from lung metastases or lymph nodes were significantly more likely to yield reactive TILs than those from gastrointestinal tumors. Even tumor size made a difference – larger tumors have a higher chance of providing viable reactive TIL cultures, presumably due to a larger starting pool of T cells and antigens [9]. These observations highlight that tumor biology (tissue of origin, burden of antigen) can affect whether one can even grow a useful TIL product.
Products can be considered suboptimal if they contain too many non-tumor-specific T cells – indeed, from a quality perspective, some teams suggest treating non-tumor-reactive T cells as “impurities” in the final product [29]. Similarly, regulatory T cells (Tregs) present in the culture are undesirable since higher Treg fractions in the infused TIL correlate with worse response rates, likely due to Tregs suppressing effector function [29, 43, 44]. In a recent study, Liu et al. reported that a higher frequency of CD3+CD4−CD8− double-negative (DN) T cells in TIL infusion products derived from nasopharyngeal carcinoma was associated with worse clinical outcomes [45]. These DN TILs, exhibiting regulatory T cell-like properties, suppress CD8+ TIL expansion and are correlated with poor prognosis. Mechanistically, DN TILs inhibit CD8+ TILs through Fas-FasL interactions and immunosuppressive signaling mediated by TGF-β and IL-10 pathways [45]. Modern protocols strive to maximize the tumor-reactive effector pool, for example, by selecting or preferentially expanding TIL subcultures that show reactivity against the tumor prior to infusion.
Infused cell dose
The total T cell dose administered has been correlated with outcomes in several studies. A meta-analysis of 112 melanoma patients treated with TILs found that responders received a significantly higher number of cells on average than non-responders. Patients infused with >50 billion cells had markedly improved overall survival compared to those receiving fewer cells [46]. These data suggest a “more is better” trend, possibly reflecting the need to overwhelm an immunosuppressive tumor with sheer force of numbers. However, the relationship is not absolute. In the pivotal C-144-01 trial that led to lifileucel’s approval, no clear association was observed between cell dose and likelihood of response [47]. This suggests that within the dose range given (10–100 billion cells) [48, 49], other qualitative factors outweighed the pure cell count in determining response, putting emphasis on cell phenotype and tumor biology. In practice, most protocols aim to infuse on the order of tens of billions of TILs, balancing potential benefit with manufacturing feasibility.
Persistence and in vivo expansion
Once infused, TILs must persist and expand within the patient to mediate sustained tumor control. The transient presence of TILs may lead to only short-lived tumor shrinkage. Thus, the ability of the transferred T cells to survive, proliferate, and populate the patient’s immune system, at least temporarily, is a key efficacy driver. In clinical studies, TIL count in the blood 1–2 weeks post-infusion is often used as a pharmacodynamic indicator, and higher levels have been associated with better clinical responses [50]. Long-term persistence of infused clones – detectable months to years later – is frequently observed in patients who achieve complete responses, suggesting those T cells continued to patrol and suppress the cancer [38]. The persistence capacity, as noted above, is tied to cell intrinsic features like memory phenotype and telomere length. It is also influenced by the host environment and supportive measures like lymphodepletion and cytokine support. Notably, the standard TIL therapy regimen includes a preparative non-myeloablative chemotherapy (typically comprising cyclophosphamide and fludarabine) [8, 9, 51] that depletes the patient’s existing lymphocytes and “makes space” for the infused TILs to engraft. This lymphodepletion also removes regulatory cells and provides a burst of homeostatic cytokines, factors known to enhance the expansion of transferred T cells. Adequate lymphodepletion is critical; however, its intensity has limits for safety and has been an area of investigation.
Exogenous cytokine support
In the classic protocol, patients receive high-dose interleukin-2 (IL-2) infusions for several days after TIL infusion to support T-cell growth and activity in vivo. IL-2 was chosen historically because it is a potent T cell growth factor, and its administration was integral to achieving TIL expansion in early trials. That said, IL-2 is far from an ideal support agent. It has a very short half-life and must be given in high doses frequently, which leads to severe toxicities (vascular leak syndrome, high fevers, hypotension, etc.) [52, 53], limiting which patients who can tolerate TIL therapy. Moreover, IL-2 not only stimulates the infused effector TILs but can also expand Tregs due to its strong activation of the IL-2 receptor alpha chain (CD25) on regulatory T cells [9]. These drawbacks motivate the search for better cytokine support strategies [21, 54]. Still, IL-2 remains a crucial component of current TIL therapy – without it, the infused cells may not expand sufficiently. The net efficacy of TIL therapy thus results from a careful choreography: the patient is lymphodepleted, a large number of partially rejuvenated T cells with tumor reactivity are infused, and IL-2 “feeds” these cells to proliferate and attack the cancer. Variations in any of these factors (cell dose, cell phenotype, specificity, persistence, and cytokine milieu) can tip the balance between a curative TIL therapy outcome and a marginal one. Ongoing trials are systematically examining each factor to optimize TIL products for different tumor settings [8, 29, 55].
Table 1 summarizes the key factors that determine TIL product quality and clinical efficacy.
Factors determining TIL product quality and clinical efficacy.
Insights from clinical trials and biomarker-guided refinements
Over the past few years, several clinical trials have provided critical insights that are shaping TIL therapy’s development. The most pivotal is the C-144-01 trial in advanced melanoma, which led to FDA approval of lifileucel. In that phase II trial, over 150 patients with metastatic melanoma refractory to anti-PD-1 and other therapies received a single infusion of lifileucel. The objective response rate was 31.4% and included durable complete responses in a subset of patients [11, 47]. Many responders maintained their remission for a year or longer after treatment, an impressive result given these patients had exhausted other options. Notably, responses were seen even in patients who had very bulky disease or poor prognostic features, although those with extensive liver or brain metastases were less likely to respond. The trial confirmed that a centrally manufactured TIL product could be delivered across multiple centers with consistent efficacy. The approval of lifileucel has energized the field, marking TIL therapy’s transition from an experimental, single-center treatment to a bona fide, FDA-recognized therapy. It also underscores the need for predictive biomarkers – even though one-third respond, the majority do not, and identifying which patients are most likely to benefit or how to enhance TILs as a medicinal product remains a priority.
The NSCLC pilot trial discussed earlier demonstrated that pre-selecting for tumor-reactive TILs can produce durable remissions in lung cancer [23]. However, such selective approaches can be technically challenging and may not always reproduce the high TIL yields seen in melanoma. A follow-up from the lung pilot, for instance, highlighted the logistical challenges in scaling up clonal selection procedures and the possibility that some patients will not have an easily expandable reactive clone. Meanwhile, in ocular melanoma (uveal melanoma), which is a low-TMB melanoma subtype that typically does not respond to checkpoint inhibitors, TIL therapy has shown early hints of efficacy when using innovative expansion protocols [58]. These disease-specific trials underscore that context matters – each tumor type may require tweaking the TIL generation or adjunct therapies.
One of the most powerful developments in refining TIL therapy comes from single-cell analyses and biomarker-driven selection. Advanced genomic tools now allow researchers to dissect TIL populations at the single-cell level, identifying which T cells are truly tumor-specific and what their phenotypes are. For example, by defining a gene expression signature (high PD-1 and TOX co-expression) associated with these neoantigen-specific cells, researchers were able to prospectively identify TILs that were >50% likely to be tumor-reactive [59]. Such findings open the door to sorting or enriching TIL products based on molecular markers rather than lengthy functional assays. Another group took a more surface-oriented approach and demonstrated that sorting TILs by CD137 (4-1BB) expression can effectively enrich for tumor-reactive, proliferative T cells in ovarian cancer and melanoma [60]. CD137 is an inducible costimulatory receptor often upregulated when T cells recognize an antigen; isolating the CD137+ subset essentially captures those T cells recently activated by tumor antigens. Indeed, a comparative analysis suggested that among various exhaustion or activation markers on TILs, CD137 positivity was the most specific marker for isolating tumor-responding T cells that could expand and attack cancer on reinfusion [61]. Leveraging this insight, researchers are refining TIL manufacturing protocols. For instance, a group at MD Anderson has experimented with adding a CD137 agonist during the initial TIL culture [62]. Other teams are investigating epigenetic modulators added during culture to preserve T cell stemness (i.e., inhibitors of certain differentiation pathways) [63].
Surface markers associated with effective TIL function are also being proposed as release criteria or quality attributes for TIL products. The presence of tissue-resident memory T cells in the infusion product, identifiable by co-expression of CD103 (an integrin that binds E-cadherin on epithelial tumor cells), has been correlated with better clinical outcomes [31]. CD103+ TILs are thought to be adept at residing in tumor tissue and mediating local immune surveillance [64, 65]. Similarly, chemokine receptors that guide T cells to tumors, such as CXCR2/3 (which bind inflammatory chemokines common in tumor microenvironments) and integrins like LFA-1 and CD49a, are considered beneficial traits for TIL homing and target engagement [66]. These findings are beginning to influence how new TIL trials are designed – for example, a product might be assessed for CD103 or CXCR3 expression as a measure of quality, or even actively sorted to enrich these subsets. In summary, a convergence of clinical trial observations and high-dimensional TIL profiling is informing a more strategic selection of TILs. Rather than infusing a random mix of tumor-infiltrating T cells, the goal is to infuse a curated population of tumor killers to maximize efficacy and consistency. However, it is important to note that biomarker-guided selection may not be universally effective due to the complex nature of TIL functionality, which may not be fully captured by any single biomarker panel. Moreover, biomarkers associated with positive outcomes in one cancer type may not necessarily yield comparable results in others.
In addition to response rates typically ranging from 30% to 60%, a major limitation of TIL therapy is disease progression occurring during the approximately month-long ex vivo TIL expansion period, which often forces patients to withdraw from clinical trials. Implementing intervening treatments tailored to patient-specific mutations during this period has been shown to improve patient retention for subsequent TIL infusion, as evidenced by a reduced dropout rate [53]. Furthermore, pre-infusion lymphodepletion regimens can lead to a decline in patient performance status, representing a significant clinical concern that warrants careful consideration [55, 67].
Table 2 summarizes key biomarkers identified through clinical trials and single-cell analyses, highlighting their role in refining TIL therapy for enhanced tumor specificity, improved efficacy, and better patient outcomes.
Biomarkers and biomarker-guided refinements in TIL therapy.
Emerging directions and future perspectives
With the first TIL therapy now approved and many ongoing trials, the field is rapidly advancing toward next-generation TIL therapies that are more potent, durable, and broadly applicable. Several key directions are being pursued, aiming to overcome current limitations.
Genetically engineered TILs
Traditional TIL products are “bulk” T cells grown from the tumor without gene modification. Researchers anticipate that genetic engineering will play a role in improving TIL functionality. One avenue is adding chimeric antigen receptors (CARs) or T-cell receptors (TCRs) to TILs to redirect them to additional tumor antigens. For example, a preclinical study introduced a CAR specific for folate receptor-alpha (an antigen enriched in ovarian cancer and some lung cancers) into TILs, coupled with CD28/4-1BB costimulatory domains, so that the engineered TILs would get a strong activation signal upon encountering tumor cells expressing that antigen [62]. This approach could endow TILs with dual specificity: their native repertoire plus the CAR. Another genetic strategy is making TILs more resistant to the immunosuppressive tumor microenvironment. One target is TGF-β signaling, a pathway that tumors exploit to suppress T cell activity. Scientists are experimenting with TILs modified to knock out or dominant-negatively inhibit the TGFβ receptor (TGFBR2), thereby rendering the TILs less susceptible to TGF-β-mediated suppression [68]. Such modified TILs might maintain function even in TGF-β-rich tumor milieu. In a first-in-human phase 1 trial, Lou et al. evaluated CRISPR-Cas9-mediated knockout of the cytokine-inducible SH2-containing protein (CISH), an intracellular immune checkpoint, in neoantigen-reactive TILs for patients with metastatic gastrointestinal cancers [69]. Among 12 patients treated, the approach was generally safe, with no severe cytokine release syndrome or neurotoxicity. One patient with microsatellite instability-high colorectal cancer achieved a complete response lasting over 21 months, while six patients experienced stable disease [69]. This study provides the first clinical evidence that targeting an intracellular checkpoint like CISH can enhance the efficacy of TIL therapy in solid tumors. Early-stage trials of engineered TILs are on the horizon, representing a fusion of TIL therapy with the gene-engineering revolution.
Cytokine enhancement and on-demand support
As discussed, systemic high-dose IL-2 is a blunt tool for supporting TILs. In the future, improved cytokine support may significantly enhance TIL therapy while reducing toxicity. One approach is to use IL-2 variants or alternative cytokines that preferentially stimulate effector T cells over Tregs. For instance, an engineered IL-2 analog (ANV149) that binds only the IL-2 receptor βγ subunits (avoiding the α-chain) is entering trials [52]. This could provide TILs with the growth signal of IL-2 without triggering Tregs or causing extreme capillary leak syndrome. IL-15, a cytokine related to IL-2, naturally supports memory T-cell maintenance and does not act on Tregs (since Tregs rely on IL-2). A phase I trial of recombinant human IL-15 given after TIL showed biological activity (e.g., expansion of NK cells as a proof of principle) but was limited by severe hypotension at higher doses [70]. Because systemic IL-15 can be toxic, a clever alternative is to have the TILs produce IL-15 themselves in a controlled way. A new product, OBX-115, uses TILs engineered with an acetazolamide-responsive gene circuit [21]. The cells contain a gene for a membrane-bound IL-15 that is kept off by a drug-controlled “switch”. When the patient takes acetazolamide (a safe oral drug), it triggers the TILs to splice and express IL-15 locally, giving themselves a proliferative boost in situ. This on-demand cytokine support could eliminate the need for systemic IL-2 or IL-15 infusions and focus the growth signal on the tumor site, minimizing side effects.
Optimizing lymphodepletion regimens
The pre-infusion conditioning regimen is another area of active research. The current standard uses cyclophosphamide and fludarabine to deplete lymphocytes. Notably, adding ultra-intensive lymphodepletion measures like total body irradiation did not improve outcomes and increased toxicity [71]. Lymphodepleting chemotherapy regimen, while effective, is highly immunosuppressive and can be rough on patients (e.g., risk of infections, organ stress) [55]. Cyclophosphamide dosing in lifileucel treatment is more than twice that used in CAR T-cell therapy, reflecting differences in lymphodepletion protocols between the two approaches [9]. Studies are investigating whether less intensive regimens might suffice [72–74]. If successful, such de-escalation could expand eligibility to older or frailer patients who currently might not tolerate TIL therapy. Additionally, many clinicians now pair or sequence TIL therapy with checkpoint inhibitor therapy – for instance, giving an anti-PD-1 antibody (like pembrolizumab) shortly after TIL infusion to block PD-1/PD-L1 interactions and keep the transferred T cells active. Lowery et al. demonstrated that adoptive transfer of neoantigen-specific TILs, selected based on their reactivity to tumor mutations (SEL-TIL), yielded enhanced tumor regressions when combined with pembrolizumab (P) [27]. Clinical responses were observed in 7.7% of patients (3 of 39) treated with SEL-TIL alone, compared to 23.5% (8 of 34) in those receiving SEL-TIL+P [27]. The rationale for adding pembrolizumab to the regimen was supported by the high expression of PD-1 on most T cells in the final infusion products. These findings underscore that PD-1 blockade effectively enhances T cell activity, persistence, and antitumor efficacy in this setting. While dose reductions of lymphodepleting chemotherapy were allowed based on clinical needs in this trial, a systematic investigation into the effects of lymphodepletion de-escalation is warranted in future studies.
Quality attributes (QAs) for TIL products
As TIL therapy becomes more standardized, there is a push to establish clear quality attributes for the final cell product that correlate with efficacy. Unlike drugs, cell therapies are living products with heterogeneous compositions, so defining the critical attributes is key to reproducibility. Recent analyses have proposed several such QAs [44, 73, 75, 76]. These include the frequency of tumor-reactive T cells in the product (measured by markers or by functional assays) [77, 78], the content of undesired cells (e.g., non-αβ T cells since high non-αβ T cell content in the product has been linked to poorer clinical outcomes) [29, 43], and the presence of beneficial phenotypes like resident memory T cells (CD103+) and homing receptors (CXCR3, LFA-1, etc.) [79] as discussed above. An insightful framework suggested that a TIL product’s identity can be captured by four functional pillars: tumor recognition, cytolytic capacity, tissue homing, and persistence [29]. Attributes contributing to each of these pillars could be quantified and used to release products or even to score/rank TIL batches. In manufacturing terms, cells that do not contribute to these desired functions can be considered impurities. For example, bystander T cells that are not tumor-reactive, or Tregs that suppress immune function, would detract from the product’s potency [27, 45, 80]. By identifying and either removing or minimizing such subsets, the next generation of TIL therapies will be more consistently potent. Regulatory agencies and producers will likely work together to define potency assays that must be met before a TIL product is administered, ensuring a higher likelihood of patient benefit.
Table 3 summarizes the emerging directions and future perspectives discussed so far.
Emerging directions in TIL therapy.
Figure 2 depicts key strategies aimed at improving the efficacy of TIL therapy by enhancing TIL product quality through advances in cell selection, manufacturing protocols, cytokine support, gene manipulation, and small-molecule agonism.
 |
Figure 2 Key strategies for enhancing the efficacy of TIL therapy. |
Conclusion
TIL therapy has transitioned from an experimental melanoma treatment to an emerging immunotherapy for a variety of solid tumors. The recent FDA approval of lifileucel validates decades of work and offers a new lifeline for patients with refractory melanoma. Yet, this achievement can be seen as just the beginning. The coming years will see TIL therapy further refined and personalized: higher-quality cells, selected for tumor reactivity and resilience; combination regimens that maintain TIL activity in vivo; and genetic enhancements that endow TILs with improved functionality.
Most patients with solid cancers, especially those with less immunogenic tumors, will likely benefit from selected or modified TIL products rather than bulk, unmodified TILs. As our understanding of the tumor-immune interplay deepens through single-cell profiling and clinical correlatives, we can better identify the lymphocyte populations capable of eradicating disease. With collaborative efforts in trial innovation and manufacturing, TIL therapy stands poised to expand its reach, bringing the curative potential witnessed in melanoma to patients with a wide range of otherwise untreatable solid tumors, including advanced/metastatic tumors.
Centralized TIL manufacturing platforms, exemplified by lifileucel, have proven effective in producing consistent, scalable products at lower cost and with greater accessibility, circumventing the infrastructure demands on individual hospitals. Expanding such centralized facilities globally will be key to broadening patient access. Nonetheless, current treatment regimens present challenges: post-infusion IL-2 administration leads to significant toxicity, and non-myeloablative chemotherapy can impair patient fitness or cause long-term complications such as myelofibrosis and bone marrow toxicity. Removing the need for these components may not only improve safety but also open avenues for retreatment in partial responders.
Combining TIL therapy with immune checkpoint blockade (ICB) offers meaningful clinical benefits, even for patients whose disease progressed on ICB-based therapy. This synergy likely stems from ICB enhancing TIL persistence and function by reversing T cell exhaustion, while checkpoint gene knockout strategies, such as CISH deletion, offer an alternative approach to intrinsically optimize TIL products. Incorporating ICB into TIL therapy more consistently may further improve outcomes in patients with solid tumors, including those refractory to standard immunotherapy. The roadmap for the future is clear – more precise, powerful, and patient-tailored TIL treatments that build on the robust foundation laid over the past thirty years.
Acknowledgments
The figures were created with BioRender.com.
Funding
This work was supported by the Florida Department of Health (FDOH) Live Like Bella awards (9LA01 and 21L10 to Q.-X.A.S.), a Florida State University (FSU) Council on Research & Creativity (CRC) grant, a grant from FSU College of Medicine Institute for Pediatric Rare Diseases, the Pfeiffer Professorship for Cancer Research in Chemistry and Biochemistry from the College of Arts & Sciences, the Diane & Michael Bruton Professor for Cancer Research, and an Endowed Chair Professorship in Cancer Research from anonymous donors (to Q.-X.A.S.).
Conflicts of interest
The authors declare no conflict of interest.
Data availability statement
This is a perspective article. No new datasets were generated. The datasets used and analyzed are published in this paper and cited from the references.
Author contribution statement
Conceptualization, D.B.S. and Q.-X.A.S.; data curation, D.B.S., M.M.G. and S.M.; formal analysis, D.B.S., M.M.G., S.M. and Q.-X.A.S.; funding acquisition, Q.-X.A.S.; investigation, D.B.S., M.M.G. and S.M.; methodology, D.B.S.; project administration, Q.-X.A.S.; resources, Q.-X.A.S.; supervision, Q.-X.A.S.; validation, D.B.S., M.M.G., S.M. and Q.-X.A.S.; visualization, D.B.S., M.M.G., and S.M.; writing—original draft preparation, D.B.S., M.M.G., and S.M.; writing—review and editing, D.B.S., M.M.G., S.M. and Q.-X.A.S. All authors have read and agreed to the published version of the manuscript.
Ethics approval
Ethical approval was not required.
References
- Haanen J, Los C, Phan GQ, et al. Adoptive cell therapy for solid tumors: current status in melanoma and next-generation therapies. American Society of Clinical Oncology Educational Book. 2024; 44(3):e431608. [Google Scholar]
- Kumar A, Watkins R, Vilgelm AE. Cell therapy with TILs: training and taming T cells to fight cancer. Frontiers in Immunology. 2021;12:690499. [Google Scholar]
- Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348(6230):62–68. [Google Scholar]
- Tang Y, Zhang AXJ, Chen G, et al. Prognostic and therapeutic TILs of cervical cancer – current advances and future perspectives. Molecular Therapy Oncolytics. 2021;22:410–430. [Google Scholar]
- Topalian SL, Rosenberg SA. Therapy of cancer using the adoptive transfer of activated killer cells and interleukin-2. Acta Haematoligca. 1987;78(Suppl 1);75–76. [Google Scholar]
- Wang S, Sun J, Chen K, et al. Perspectives of tumor-infiltrating lymphocyte treatment in solid tumors. BMC Medicine. 2021;19:140. [Google Scholar]
- Muul LM, Spiess PJ, Director EP, et al. Identification of specific cytolytic immune responses against autologous tumor in humans bearing malignant melanoma. Journal of immunology (Baltimore, Md.: 1950). 1987;138(3):989–995. [Google Scholar]
- Albarrán Fernández V, Ballestín Martínez P, Stoltenborg Granhøj J, et al. Biomarkers for response to TIL therapy: a comprehensive review. Journal for ImmunoTherapy of Cancer. 2024;12(3):e008640. [Google Scholar]
- Shoushtari AN, Powell DJ. Tumor-infiltrating lymphocyte therapy for melanoma and other solid tumors: looking back, yet moving forward. Transplantation and Cellular Therapy. 2025;31(3):S581–S590. [Google Scholar]
- Parums DV. First regulatory approval for adoptive cell therapy with autologous tumor-infiltrating lymphocytes (TILs) – Lifileucel (Amtagvi). Medical Science Monitor. 2024;30:e944927. [Google Scholar]
- Chesney J, Lewis KD, Kluger H, et al. Efficacy and safety of lifileucel, a one-time autologous tumor-infiltrating lymphocyte (TIL) cell therapy, in patients with advanced melanoma after progression on immune checkpoint inhibitors and targeted therapies: pooled analysis of consecutive cohorts of the C-144-01 study. Journal for ImmunoTherapy of Cancer. 2022;10(12):e005755. [Google Scholar]
- Zhu Y, Zhou J, Zhu L, et al. Adoptive tumor infiltrating lymphocytes cell therapy for cervical cancer. Human Vaccines and Immunotherapeutics. 2022;18(5):2060019. [Google Scholar]
- Piroozkhah M, Gholinezhad Y, Piroozkhah M, et al. The molecular mechanism of actions and clinical utilities of tumor infiltrating lymphocytes in gastrointestinal cancers: a comprehensive review and future prospects toward personalized medicine. Frontiers in Immunology. 2023;14:1298891. [Google Scholar]
- Yang J, Ding X, Fang Z, et al. Association of CD8+ TILs co-expressing granzyme A and interferon-γ with colon cancer cells in the tumor microenvironment. BMC Cancer. 2024;24(1):869. [Google Scholar]
- Betof Warner A, Corrie PG, Hamid O. Tumor-infiltrating lymphocyte therapy in melanoma: facts to the future. Clinical Cancer Research. 2023;29(10):1835–1854. [Google Scholar]
- Turcotte S, Donia M, Gastman B, et al. Art of TIL immunotherapy: SITC’s perspective on demystifying a complex treatment. Journal for ImmunoTherapy of Cancer. 2025;13(1):e010207. [Google Scholar]
- Zhao Y, Deng J, Rao S, et al. Tumor infiltrating lymphocyte (TIL) therapy for solid tumor treatment: progressions and challenges. Cancers. 2022;14(17):4160. [Google Scholar]
- Mullinax JE, Hall M, Prabhakaran S, et al. Combination of ipilimumab and adoptive cell therapy with tumor-infiltrating lymphocytes for patients with metastatic melanoma. Frontiers in Oncology. 2018;8:44. [Google Scholar]
- Vafaei S, Zekiy AO, Khanamir RA, et al. Combination therapy with immune checkpoint inhibitors (ICIs); a new frontier. Cancer Cell International. 2022;22:1–27. [Google Scholar]
- Chamberlain CA, Bennett EP, Kverneland AH, et al. Highly efficient PD-1-targeted CRISPR-Cas9 for tumor-infiltrating lymphocyte-based adoptive T cell therapy. Molecular Therapy Oncolytics. 2022;24:417–428. [Google Scholar]
- Amaria RN, McQuade JL, Davies MA, et al. OBX-115, an interleukin 2 (IL2)-sparing engineered tumor-infiltrating lymphocyte (TIL) cell therapy, in patients (pts) with immune checkpoint inhibitor (ICI)-resistant unresectable or metastatic melanoma. Journal of Clinical Oncology. 2024;42:9515–9515. [Google Scholar]
- Peng W, Ye Y, Rabinovich BA, et al. Transduction of tumor-specific T cells with CXCR2 chemokine receptor improves migration to tumor and antitumor immune responses. Clinical Cancer Research. 2010;16(22):5458–5468. [Google Scholar]
- Creelan BC, Wang C, Teer JK, et al. Tumor-infiltrating lymphocyte treatment for anti-PD-1-resistant metastatic lung cancer: a phase 1 trial. Nature Medicine. 2021;27(8):1410–1418. [Google Scholar]
- Stevanović S, Draper LM, Langhan MM, et al. Complete regression of metastatic cervical cancer after treatment with human papillomavirus-targeted tumor-infiltrating T cells. Journal of Clinical Oncology. 2015;33(14):1543–1550. [Google Scholar]
- Stevanović S, Pasetto A, Helman SR, et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science. 2017;356(6334):200–205. [Google Scholar]
- Stevanović S, Helman SR, Wunderlich JR, et al. A phase II study of tumor-infiltrating lymphocyte therapy for human papillomavirus-associated epithelial cancers. Clinical Cancer Research. 2019;25(5):1486–1493. [Google Scholar]
- Lowery FJ, Goff SL, Gasmi B, et al. Neoantigen-specific tumor-infiltrating lymphocytes in gastrointestinal cancers: a phase 2 trial. Nature Medicine. 2025;31:1994–2003. [Google Scholar]
- Tran E, Turcotte S, Gros A, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014;344(6184):641–645. [Google Scholar]
- Lievense JJ, Nijenhuis C, Jedema I, et al. Defining the quality attributes for tumor-infiltrating lymphocyte medicinal products. Transplantation and Cellular Therapy. 2025;31(3):S610–S625. [Google Scholar]
- Radvanyi LG, Bernatchez C, Zhang M, et al. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clinical Cancer Research. 2012;18(24):6758–6770. [Google Scholar]
- Kverneland AH, Chamberlain CA, Borch TH, et al. Adoptive cell therapy with tumor-infiltrating lymphocytes supported by checkpoint inhibition across multiple solid cancer types. Journal for ImmunoTherapy of Cancer. 2021;9(10):e003499. [Google Scholar]
- Hall MS, Teer JK, Yu X, et al. Neoantigen-specific CD4+ tumor-infiltrating lymphocytes are potent effectors identified within adoptive cell therapy products for metastatic melanoma patients. Journal for ImmunoTherapy of Cancer. 2023;11(10):e007288. [Google Scholar]
- Oliveira G, Stromhaug K, Klaeger S, et al. Phenotype, specificity and avidity of antitumour CD8+ T cells in melanoma. Nature. 2021;596(7870):119–125. [Google Scholar]
- Simoni Y, Becht E, Fehlings M, et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. 2018;557(7706):575–579. [CrossRef] [PubMed] [Google Scholar]
- Jansen CS, Prokhnevska N, Master VA, et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature. 2019;576:465–470. [Google Scholar]
- Sade-Feldman M, Yizhak K, Bjorgaard SL, et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell. 2018;175(4):998–1013.e20. [CrossRef] [PubMed] [Google Scholar]
- Huang J, Khong HT, Dudley ME, et al. Survival, persistence, and progressive differentiation of adoptively transferred tumor-reactive T cells associated with tumor regression. Journal of Immunotherapy. 2005;28(3):258–267. [Google Scholar]
- Rosenberg SA, Yang JC, Sherry RM, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clinical Cancer Research. 2011;17(13):4550–4557. [Google Scholar]
- Zhou J, Shen X, Huang J, et al. Telomere length of transferred lymphocytes correlates with in vivo persistence and tumor regression in melanoma patients receiving cell transfer therapy. Journal of immunology. 2005;175(10):7046–7052. [Google Scholar]
- Gattinoni L, Klebanoff CA, Palmer DC, et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. Journal of Clinical Investigation. 2005;115(6):1616–1626. [Google Scholar]
- Tran KQ, Zhou J, Durflinger KH, et al. Minimally cultured tumor-infiltrating lymphocytes display optimal characteristics for adoptive cell therapy. Journal of Immunotherapy. 2008;31(8):742–751. [Google Scholar]
- Donia M, Junker N, Ellebaek E, et al. Characterization and comparison of “Standard” and “young” tumour‐infiltrating lymphocytes for adoptive cell therapy at a Danish Translational research institution. Scandinavian Journal of Immunology. 2012;75(2):157–167. [Google Scholar]
- Chiffelle J, Barras D, Pétremand R, et al. Tumor-reactive T cell clonotype dynamics underlying clinical response to TIL therapy in melanoma. Immunity, 2024;51(10):2466–2482. [Google Scholar]
- Zhou X, Wu J, Duan C, et al. Retrospective analysis of adoptive TIL therapy plus anti‐PD1 therapy in patients with chemotherapy‐resistant metastatic osteosarcoma. Journal of Immunology Research. 2020;2020(1):7890985. [Google Scholar]
- Liu XF, Song B, Sun CB, et al. Tumor-infiltrated double-negative regulatory T cells predict outcome of T cell-based immunotherapy in nasopharyngeal carcinoma. Cell Reports Medicine. 2025;6(5):102096. [Google Scholar]
- Dafni U, Michielin O, Lluesma SM, et al. Efficacy of adoptive therapy with tumor-infiltrating lymphocytes and recombinant interleukin-2 in advanced cutaneous melanoma: a systematic review and meta-analysis. Annals of Oncology. 2019;30(12):1902–1913. [Google Scholar]
- Sarnaik AA, Hamid O, Khushalani NI, et al. Lifileucel, a tumor-infiltrating lymphocyte therapy, in metastatic melanoma. Journal of Clinical Oncology. 2021;39(24):2656–2666. [Google Scholar]
- Nguyen LT, Yen PH, Nie J, et al. Expansion and characterization of human melanoma tumor-infiltrating lymphocytes (TILs). PLoS One. 2010;5(11):e13940. [Google Scholar]
- van den Berg JH, Heemskerk B, van Rooij N, et al. Tumor infiltrating lymphocytes (TIL) therapy in metastatic melanoma: boosting of neoantigen-specific T cell reactivity and long-term follow-up. Journal for ImmunoTherapy of Cancer. 2020;8(2):e000848. [Google Scholar]
- Besser MJ, Itzhaki O, Ben‐Betzalel G, et al. Comprehensive single institute experience with melanoma TIL: Long term clinical results, toxicity profile, and prognostic factors of response. Molecular Carcinogenesis. 2020;59(7):736–744. [Google Scholar]
- Albarrán V, San Román M, Pozas J, et al. Adoptive T cell therapy for solid tumors: current landscape and future challenges. Frontiers in Immunology. 2024;15:1352805. [Google Scholar]
- Joerger M, Calvo E, Laubli H, et al. Phase 1 first-in-human dose-escalation study of ANV419 in patients with relapsed/refractory advanced solid tumors. Journal for ImmunoTherapy of Cancer. 2023;11(11):e007784. [Google Scholar]
- Sarnaik AA, Hwu P, Mulé JJ, et al. Tumor-infiltrating lymphocytes: a new hope. Cancer Cell. 2024;42(8):1315–1318. [Google Scholar]
- Zhang L, Morgan RA, Beane JD, et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clinical Cancer Research. 2015;21(10):2278–2288. [Google Scholar]
- Betof Warner A, Hamid O, Komanduri K, et al. Expert consensus guidelines on management and best practices for tumor-infiltrating lymphocyte cell therapy. Journal for ImmunoTherapy of Cancer. 2024;12(2):e008735. [Google Scholar]
- Heimann A. Elongated T cell expansion by utilizing IL7 and IL15 leads to increased T cell yield while preserving Tcm/Tscm characteristics feasible for adoptive T cell therapy. Germany: Freie Universitaet Berlin; 2020. [Google Scholar]
- Pilipow K, Roberto A, Roederer M, et al. IL15 and T-cell stemness in T-cell-based cancer immunotherapy. Cancer Research. 2015;75(24):5187–5193. [Google Scholar]
- Leonard-Murali S, Kammula US. Optimizing TIL therapy for uveal melanoma: lessons learned and unlearned from cutaneous melanoma. Immunotherapy. 2025;17(4):283–291. [Google Scholar]
- Lowery FJ, Krishna S, Yossef R, et al. Molecular signatures of antitumor neoantigen-reactive T cells from metastatic human cancers. Science. 2022;375(6583):877–884. [Google Scholar]
- Ye Q, Song DG, Poussin M, et al. CD137 accurately identifies and enriches for naturally occurring tumor-reactive T cells in tumor. Clinical Cancer Research. 2014;20(1):44–55. [Google Scholar]
- Eiva MA, Omran DK, Chacon JA, et al. Systematic analysis of CD39, CD103, CD137, and PD‐1 as biomarkers for naturally occurring tumor antigen‐specific TILs. European Journal of Immunology. 2022;52(1):96–108. [Google Scholar]
- Tavera RJ, Forget MA, Kim YU, et al. Utilizing T-cell activation signals 1, 2, and 3 for tumor-infiltrating lymphocytes (TIL) expansion: the advantage over the sole use of interleukin-2 in cutaneous and uveal melanoma. Journal of Immunotherapy. 2018;41(9):399–405. [Google Scholar]
- Matsueda S, Chen L, Li H, et al. Recent clinical researches and technological development in TIL therapy. Cancer Immunology, Immunotherapy. 2024;73(11):232. [Google Scholar]
- Duhen T, Duhen R, Montler R, et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nature Communications. 2018;9(1):2724. [Google Scholar]
- Kim Y, Shin Y, Kang GH. Prognostic significance of CD103+ immune cells in solid tumor: a systemic review and meta-analysis. Scientific Reports. 2019;9(1):3808. [Google Scholar]
- Franciszkiewicz K, Le Floch A, Boutet M, et al. CD103 or LFA-1 engagement at the immune synapse between cytotoxic T cells and tumor cells promotes maturation and regulates T-cell effector functions. Cancer Research. 2013;73(2):617–628. [Google Scholar]
- Li D, Chen C, Li J, et al. A pilot study of lymphodepletion intensity for peripheral blood mononuclear cell-derived neoantigen-specific CD8+ T cell therapy in patients with advanced solid tumors. Nature Communications. 2023;14(1):3447. [Google Scholar]
- Fix SM, Forget MA, Sakellariou-Thompson D, et al. CRISPR-mediated TGFBR2 knockout renders human ovarian cancer tumor-infiltrating lymphocytes resistant to TGF-β signaling. Journal for ImmunoTherapy of Cancer. 2022;10(7):e003750. [Google Scholar]
- Lou E, Choudhry MS, Starr TK, et al. Targeting the intracellular immune checkpoint CISH with CRISPR-Cas9-edited T cells in patients with metastatic colorectal cancer: a first-in-human, single-centre, phase 1 trial. Lancet Oncology. 2025;26(5):559–570. [Google Scholar]
- Conlon KC, Lugli E, Welles HC, et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. Journal of Clinical Oncology. 2015;33(1):74–82. [Google Scholar]
- Goff SL, Dudley ME, Citrin DE, et al. Randomized, prospective evaluation comparing intensity of lymphodepletion before adoptive transfer of tumor-infiltrating lymphocytes for patients with metastatic melanoma. Journal of Clinical Oncology. 2016;34(20):2389–2397. [Google Scholar]
- Guo J, Wang C, Luo N, et al. IL-2-free tumor-infiltrating lymphocyte therapy with PD-1 blockade demonstrates potent efficacy in advanced gynecologic cancer. BMC Medicine. 2024;22(1):207. [Google Scholar]
- Lickefett B, Chu L, Ortiz-Maldonado V, et al. Lymphodepletion – an essential but undervalued part of the chimeric antigen receptor T-cell therapy cycle. Frontiers in Immunology. 2023;14:1303935. [Google Scholar]
- Verdegaal EM, Verpoorte ALC, van der Kooij MK, et al. Effective TIL therapy for patients with checkpoint-resistant melanoma without lymphodepleting regimens requires IFNα. Clinical Cancer Research. 2025;31:2628–2638. [Google Scholar]
- Friese C, Harbst K, Borch TH, et al. CTLA-4 blockade boosts the expansion of tumor-reactive CD8+ tumor-infiltrating lymphocytes in ovarian cancer. Scientific Reports. 2020;10(1):3914. [Google Scholar]
- Martinez-Usatorre A, Carmona SJ, Godfroid C, et al. Enhanced phenotype definition for precision isolation of precursor exhausted tumor-infiltrating CD8 T cells. Frontiers in Immunology. 2020;11:340. [Google Scholar]
- Kristensen NP, Heeke C, Tvingsholm SA, et al. Neoantigen-reactive CD8+ T cells affect clinical outcome of adoptive cell therapy with tumor-infiltrating lymphocytes in melanoma. Journal of Clinical Investigation. 2022;132(2):e150535. [Google Scholar]
- Levi ST, Copeland AR, Nah S, et al. Neoantigen identification and response to adoptive cell transfer in anti-PD-1 naïve and experienced patients with metastatic melanoma. Clinical Cancer Research. 2022;28(14):3042–3052. [Google Scholar]
- Yuhas A, Dean J, Erickson T, et al. Effect of a novel expansion process on tumor-infiltrating lymphocyte (TIL) polyfunctionality, cytotoxicity, and expansion, while preserving cells in a less differentiated and more stem-like phenotype. Journal of Clinical Oncology. 2023;41:2542–2542. [Google Scholar]
- Turcotte S, Gros A, Hogan K, et al. Phenotype and function of T cells infiltrating visceral metastases from gastrointestinal cancers and melanoma: implications for adoptive cell transfer therapy. Journal of Immunology. 2013;191(5):2217–2225. [Google Scholar]
Cite this article as: MM, Martinez S & Sang Q-XA. Tumor-infiltrating lymphocyte therapy: therapeutic advances and prospects. Visualized Cancer Medicine. 2025, 6, 9. https://doi.org/10.1051/vcm/2025010.
All Tables
All Figures
|
Figure 1 Overview of tumor-infiltrating lymphocyte (TIL) therapy. Tumors are surgically resected from patients, and tumor-infiltrating lymphocytes (TILs) are isolated and expanded ex vivo. These cells may either be infused without modification or genetically engineered. Engineered TILs include CAR (chimeric antigen receptor) TILs, checkpoint protein knockout (KO) TILs (e.g., programmed cell death protein 1 (PD-1) KO), and TILs expressing cytokine (e.g., interleukin-15 or IL-15) or chemokine receptor (e.g., CXC chemokine receptor 2 or CXCR2) to enhance persistence, function, or trafficking. Upon reinfusion into the patient, activated TILs specifically home to and target cancer cells, mediating effective tumor cell killing and potentially leading to durable cancer regression. |
| In the text | |
|
Figure 2 Key strategies for enhancing the efficacy of TIL therapy. |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.